

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Mehta A, Beck M, Sunder-Plassmann G, editors. Fabry Disease: Perspectives from 5 Years of FOS. Oxford: Oxford PharmaGenesis; 2006.

Fabry disease can be diagnosed in affected males by demonstrating a deficiency of α-galactosidase A in plasma and leukocytes. However, the enzymatic assay is unreliable for the detection of carriers, who can be detected reliably only by mutational analysis. All classic hemizygotes and over 90% of heterozygotes have an elevated level of urinary globotriaosylceramide (Gb3 ). For male patients with lower than normal α-galactosidase activity or with an atypical clinical presentation, measurement of urinary Gb3 and sequencing of the whole GLA gene must be carried out.
Biochemistry and structure of α-galactosidase A
A deficiency of α-galactosidase (ceramide-trihexosidase) was first shown to be the enzymatic defect in Fabry disease in 1967 by Brady and colleagues using radiolabelled globotriaosylceramide (Gb3) as the substrate [1]. Subsequently, Kint [2] demonstrated that the α-galactosidase could act on the synthetic substrates p-nitrophenyl–α-d-galactoside and 4-methylumbelliferyl–α-d-galactoside and was specific for the α-anomeric galactosidic linkage. The introduction of synthetic substrates facilitated the assay of α-galactosidase activity for the diagnosis of Fabry disease [3] and the characterization and purification of the enzyme [4]. Investigation of α-galactosidase activity in normal individuals and residual activity in patients with Fabry disease revealed that there were two α-galactosidase isoenzymes, A and B, with activity towards the synthetic substrates [5]. α-Galactosidase A, which is thermolabile, accounts for most of the activity in normal tissues and is deficient in patients with Fabry disease. In contrast, α-galactosidase B, which is heat stable, accounts for the residual activity in patients with Fabry disease, indicating that the two activities are genetically distinct. The purified enzymes have many similar physicochemical properties, but can be separated on the basis of charge. Antibodies raised against one do not cross-react with the other, supporting their genetic difference [4]. In fact, α-galactosidase B is an α-N-acetylgalactosaminidase that acts on natural substrates with terminal α-N-acetylgalactosaminyl residues [6, 7] and is defective in Schindler disease [8]. α-Galactosidase A does not catalyse the hydrolysis of the natural substrates of α-galactosidase B, whereas α-galactosidase B may act on some natural substrates with terminal α-galactosyl residues [9]. Therefore, as both enzymes act on synthetic α-galactoside substrates, a specific inhibitor of α-galactosidase B, α-N-acetylgalactosamine, is added to the assay mixture when measuring the α-galactosidase activity for diagnosing Fabry disease [10]. The genes encoding α-galactosidase A (GLA) and α-galactosidase B (NAGA) are localized on chromosomes Xq22.1 and 22q13, respectively, confirming that the two enzymes are genetically distinct. Cloning of the two genes suggested that they were evolutionarily related, with a predicted 46.9% identity of the amino acid sequence at the complementary DNA (cDNA) level and great similarity in the organization of a large section of the genes [11].
α-Galactosidase A is a glycoprotein and is synthesized as a precursor of 50 kDa, which is processed to a mature lysosomal form of 46 kDa following removal of a signal peptide during its transport to the lysosome via the mannose-6-phosphate (M6P) pathway [12]. The active enzyme is a homodimer that requires saposin B to act on its natural substrates in vivo [13]. As in Fabry disease, patients with a genetic deficiency of saposin B also accumulate the substrates for α-galactosidase A [14]. The predominant storage product in Fabry disease, and therefore the major natural substrate for α-galactosidase A, is Gb3 [Gal(α1→4)Gal(β1→4)Glc(β1→1′)Cer], also called CTH or GL-3. The hydrolysis of Gb3 in vitro requires the addition of a detergent, usually sodium taurocholate. Another natural substrate is galabiosylceramide, [Gal(α1→4) Gal(β1→1′)Cer] or Ga2, which also accumulates in Fabry disease. Both of these neutral glycolipids consist of families of isoforms arising from heterogeneity in the fatty acid component of the ceramide [15]. Two other glycolipid substrates, which can accumulate in Fabry disease depending on the blood group and secretor status of the patient, are the blood group B and B1 glycosphingolipids, which have terminal α-galactosyl residues [9, 16]. α-Galactosidase A is a typical lysosomal acid hydrolase with an optimal activity towards natural and synthetic substrates at pH 3.8–4.6 [17]. The Michaelis constant (Km) values for synthetic substrates are approximately one order of magnitude higher than for natural substrates.
Cloning of the GLA gene has permitted the production of recombinant human α-galactosidase A for enzyme replacement therapy (ERT) in Fabry disease [18, 19] and for structural analysis of the enzyme. Currently, two preparations are available, one of which (agalsidase alfa; Replagal®, TKT Europe AB) is produced in a continuous human cell line and the other (agalsidase beta; Fabrazyme®, Genzyme Corp.) in Chinese hamster ovary cells. Both proteins have the same amino acid sequence [20] but differ in their N-linked glycosylation because they are produced in different cell lines [21]. Three out of the four potential N-glycosylation sites are occupied in both preparations [22] but agalsidase beta has higher levels of sialic acid and M6P on its N-linked glycans than agalsidase alfa [21]. The rate of uptake of both preparations into fibroblasts via the M6P receptor is very similar and leads to the dispersal of accumulated Gb3 [20]. The distribution of the two enzymes in knockout Fabry mice was comparable after intravenous administration. The enzymes have similar kinetics towards synthetic substrates [21] and show complete cross-reactivity [23].
The three-dimensional structure of recombinant human α-galactosidase A (agalsidase alfa) has been determined by X-ray crystallography at a resolution of 3.25 Å (Figure 1) [24]. It is a homodimeric molecule, with each monomer made up of two domains, one containing the active site and the other a β-sandwich of anti-parallel β-strands at the C-terminal. Co-crystallization with the product, galactose, revealed multiple interactions between the monosaccharide and parts of the first domain, confirming the location of the active site and suggesting that two aspartic acid residues could act as a nucleophile and acid/base in the catalytic mechanism. The distance between the two active sites is approximately 50 Å in both the free and liganded enzymes, suggesting that there is no cooperation between the sites. Comparison of the three-dimensional structures of α-galactosidase A and α-galactosidase B (α-N-acetylgalactosaminidase) confirms the structural similarity between the two enzymes [24, 25]. The absence of an 'N-acetyl recognition loop' in α-galactosidase A offers an explanation for the difference in substrate specificity. The X-ray crystal structures of free human α-galactosidase expressed in yeast and as a complex with an inhibitor, N-(N-benzyloxycarbonyl-6-aminohexyl)-α-d-galactopyranoside, have also been reported [26]. The three-dimensional structures have been used to understand the effects of mutations on the function of the enzyme (see Chapters 33 and 34).

Practical aspects of enzyme determination
α-Galactosidase A activity can easily be measured in plasma and leukocytes using the synthetic substrate 4-methylumbelliferyl-α-d-galactopyranoside [2, 3]. The effects of pH, temperature, inhibitors, anticoagulants and erythrocyte contamination on the activity of α-galactosidase A in plasma, serum, urine and leukocytes have been investigated [3]. These findings were taken into account in the development of assays and establishment of reference ranges for α-galactosidase A in plasma and leukocytes, which have been used in our laboratory for over 30 years [27, 28] (Figures 2 and 3). Classically affected hemizygotes have very low or undetectable enzyme activity but some hemizygotes, for example those with the N215S mutation, may have higher residual activity in plasma and/or leukocytes. It is therefore important to confirm the mutation in these individuals. The α-galactosidase A activity in affected females can range from the low level found in affected males to well into the normal range, possibly due to skewed X-inactivation. Therefore, heterozygotes cannot be reliably defined by enzymatic analysis.
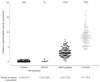
Figure 2
Levels of plasma α-galactosidase in male hemizygotes with classic Fabry disease and with the N215S mutation, in female heterozygotes and in healthy control individuals. Numbers of subjects are given in parentheses.
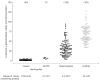
Figure 3
Levels of leukocyte α-galactosidase in male hemizygotes with classic Fabry disease and with the N215S mutation, in female heterozygotes and in healthy control individuals. Numbers of subjects are given in parentheses.
Pitfalls in plasma assays
As 100 μl of plasma/serum are used in the assay, a relatively strong acetate buffer is needed to maintain the incubation pH of 4.8. It is essential to add the inhibitor N-acetyl-galactosamine (final concentration, 100 mmol/l) to inhibit the α-N-acetylgalactosaminidase [10], which can account for up to 20% of the α-galactosidase activity in plasma. Pre-incubation of plasma at 37°C for 2 hours before the assay, decreased the α-galactosidase activity by approximately 80% [3]. In contrast, the activity was essentially unchanged when plasma was preheated at 25°C. To minimize the effect of denaturation and to ensure measurable hydrolysis of the substrate, assays are therefore performed at 30°C. Although there is no significant difference between the activity in serum or plasma collected in tubes with ethylenediamine tetraacetic acid, heparin or acid-citrate-dextrose as anticoagulants, substances released from erythrocytes, for example haemoglobin, can significantly reduce fluorescence. This is corrected for by having a separate standard, standard blank and substrate blank for each sample assayed. The blank readings may vary considerably from sample to sample. The results from lipaemic plasma may be suspect because, depending on the severity of lipaemia, the solution may not clear after addition of the stopping reagent and this can affect the fluorescence. As with all diagnostic samples, another enzyme (e.g. β-hexosaminidase) should be assayed in the plasma/serum sample to ensure the viability of the sample.
Pitfalls in leukocyte assays
There are fewer pitfalls in the leukocyte assay than in the plasma assay. To solubilize the activity fully, leukocytes are homogenized in 0.5% sodium cholate [28], or frozen and thawed ten times [3], and the supernatant is used in the enzyme assays. The α-galactosaminidase activity accounts for only 5–10% of the total α-galactosidase activity in leukocytes, so the addition of N-acetylgalactosamine to the assay is not as important as in the plasma assay. However, it is recommended that it is included. As with plasma, haemoglobin contamination can quench the fluorescence but, again, this is usually not a problem, as contaminated erythrocytes can be removed by a haemolysis step in the leukocyte preparation. Another enzyme (e.g. β-galactosidase) should be assayed in the same extract to ensure the viability of the sample.
Male patients with residual α-galactosidase activity
There have been many reports of men with decreased but not absent α-galactosidase activity, who do not show the classic clinical phenotype of Fabry disease. Some of these patients have clinical variants, with the initial presenting symptoms restricted to a particular organ, for example the heart or kidneys, and such variants are often associated with particular mutations [5]. It is important to note that many of these mutations have also been found in patients with the classic phenotype, indicating that other factors affect the phenotype. The coding sequence variant, D313Y, which has approximately 60% of wild-type activity in vitro and decreased activity at neutral pH, leading to low plasma α-galactosidase activity, is not disease causing [29, 30]. It has an incidence of 0.45% in normal X chromosomes and has been found in combination with several disease causing missense mutations. The discovery of this pseudodeficiency allele emphasizes the need to sequence the GLA gene completely in the index case of newly diagnosed families with Fabry disease and to express novel missense mutations. Patients with an atypical clinical presentation and residual α-galactosidase activity should be investigated thoroughly before making a definitive diagnosis. This should include characterization of the residual α-galactosidase activity, measurement of urinary Gb3 and sequencing of the whole GLA gene.
Prenatal diagnosis
Measurement of α-galactosidase A activity in cultured amniotic cells [31] and directly in chorionic villi [32] has been used for the prenatal diagnosis of Fabry disease. More recently, karyotype analysis followed by enzyme analysis and/or mutational analysis of a male fetus has become the usual procedure. With the advent of ERT, however, there are now very few requests for prenatal diagnosis of Fabry disease.
The availability of ERT has emphasized the importance of early diagnosis of Fabry disease after birth, even in newborns. Deficiency of α-galactosidase can be demonstrated in activity eluted from a dried blood spot collected on filter paper [33] (see Chapter 17).
Measurement of storage products
The accumulation of the major storage product, in various tissues is the Gb3, hallmark of Fabry disease. Many methods have therefore been developed to detect and measure Gb3, including thin-layer chromatography [34], gas-liquid chromatography [35], high-performance liquid chromatography [36] and an enzyme-linked immunosorbent assay using the verotoxin B subunit [37]. Glycosphingolipids can be detected and readily identified by various forms of mass spectrometry [38–41]. Initially, derivatization was necessary to prevent decomposition and is still used to achieve sensitivity. The advent of softer ionization methods and tandem mass spectrometry has permitted the analysis of small amounts of non-derivatized neutral and acidic glycosphingolipids, including detection of the isoforms due to heterogeneity in the sphingosine and fatty acid components. However, until recently, quantitative determination was handicapped by the lack of suitable internal standards. The synthesis of the novel internal standards C-17-Gb3 [42],[d4]C16-and [d47]C24-isoforms of Gb3 and Ga2 [43] and [d35]C18-Gb3 [44] has overcome this problem. These standards and the development of simpler extraction and on-line purification procedures have permitted the rapid, automated and quantitative determination of non-derivatized Gb3 in plasma and urine by liquid chromatography in conjunction with electrospray ionization tandem mass spectrometry [42, 44, 45]. Sonication of whole urine [46] or dilution of whole urine [45] was found to give reliable results without the need to separate the sediment and supernatant. The concentration of total Gb3 is obtained by summating the concentrations of the individual isoforms of Gb3 detected by using the mass spectrometer in the multiple-reaction monitoring mode [15, 42, 44–48]. This methodology has been used to establish reference ranges for total Gb3 in plasma and urine from Fabry hemizygotes, heterozygotes and normal controls [15, 46, 47] (Table 1 and Figures 4 and 5).
Table 1
Proportion of hemizygous male and heterozygous female patients with Fabry disease (with or without the N215S mutation) with elevated levels of globotriaosylceramide (Gb3 ) in plasma and urine.
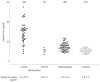
Figure 4
Levels of plasma globotriaosylceramide (Gb3 ) in male hemizygotes with classic Fabry disease and with the N215S mutation, in female heterozygotes and in healthy control individuals. Numbers of subjects are given in parentheses.
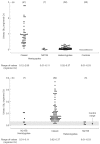
Total Gb3 was elevated in the plasma of 44 out of 48 classic hemizygotes studied, but not in some mildly affected male patients and those with the N215S mutation. Eight out of nine boys (ages ranging from 6 to 17 years) had elevated levels of Gb3. Only 42% of female heterozygotes and only one of four N215S heterozygotes had elevated plasma levels of Gb3. In contrast, all of the classic male hemizygotes and three of the seven patients with the N215S mutation had elevated urinary Gb3 levels. The proportion of heterozygotes with elevated urinary Gb3 was 95%, and all but one of the heterozygotes with a normal level of urinary Gb3 were symptomatic. The urinary Gb3 level was within the normal range for the N215S heterozygotes. Thus, urinary Gb3 is a better marker of Fabry disease than plasma Gb3, but does not detect some heterozygotes [47, 48]. Measurement of urinary and plasma Gb3 by tandem mass spectrometry has been used to monitor ERT [45–50].
Attempts have been made to identify heterozygotes by obtaining a urinary lipid profile by tandem mass spectrometry [51] but this did not detect all female heterozygotes. Fingerprints of urinary sediment glycosphingolipids obtained using matrix-assisted laser desorption ionization time-of-flight mass spectrometry [52] indicated that there are significant differences in the composition of the glycosphingolipids amongst young, adult and atypical hemizygous and heterozygous patients. It is possible that these changes could be used to assess the severity, progress and pathophysiology of Fabry disease.
Methods of genetic diagnosis
The GLA gene, which is located at Xq22.1, has been fully characterized [5, 53, 54]. It is 12 kilobases (kb) in length and contains seven exons, which have consensus intron/exon splicing sequences. There are several possible regulatory elements in the 5′-flanking region and an unmethylated CpG-rich island upstream of the initiation codon, which is characteristic of a housekeeping gene. The gene contains 12 Alu repetitive elements, about 20% of the gene. The mRNA (1.45 kb), which is unusual in lacking a 3′-untranslated sequence, encodes a protein of 429 amino acids, including the N-terminal signal peptide of 31 amino acids. Another unusual feature is the presence of three polymorphisms with a combined frequency of 10% in the 60 base pair untranslated sequence before the initiation codon in the first exon [55]. One of these, −30G>A, which is found in 0.5% of normal individuals, gives rise to elevated plasma α-galactosidase activity [56]. In contrast, the pseudodeficiency allele (D313Y) lowers plasma α-galactosidase activity [29, 30].
Mutations in the GLA gene have been identified using a variety of strategies. Large gene rearrangements, which account for only 3–4% of the mutations, can be detected by Southern blot hybridization using full-length cDNA as the probe [57] or by multiplex polymerase chain reaction (PCR) of the gene in four fragments [58]. The sizes of the seven exons, 92–291 base pairs in length, lend themselves to amplification by PCR and detection of sequence changes by mutation screening methods. Single base changes and small deletions and insertions can be detected by single-strand conformation polymorphism analysis [55], chemical cleavage of mismatches with fluorescent detection [59] or denaturing high-performance liquid chromatography (DHPLC) [60], followed by sequencing of exons to identify the precise mutation. Direct sequencing of the entire coding sequence and flanking regions in both directions has also been used to identify mutations [61]. Mutations in the conserved 5′-splice donor sites and 3′-splice acceptor sites should be investigated further by northern blotting to define the effect on RNA processing.
Once the mutation in the index case has been identified, a specific test for the mutation is developed for investigation of family members. If the mutation alters an existing restriction enzyme site in the genomic DNA, the mutation can be detected by re-amplification of the DNA fragment and digestion with the enzyme. If the mutation does not alter a restriction site, a restriction site can be created using a primer containing an altered base (amplification-created restriction site) [62]. A specific mutation can also be rapidly detected by DHPLC analysis of the amplified fragment that contains the mutation and confirmed by sequencing [60]. Direct sequencing of the appropriate amplified fragment is a feasible strategy with the development of rapid and sensitive automated sequencers. Known single-point and small deletions can also be detected by a rapid, fluorophore probe-based technique that does not require gel electrophoresis [63]. Two probes with different fluorophores are designed to bind one base pair apart to a PCR product, with one probe covering the mutation. The proximity of the probes leads to fluorescence resonance energy transfer and emission of fluorescence of a specific wavelength. When such complexes are subjected to melting, the specific emission is lost and a melting curve can be recorded using a LightCycler (Roche Diagnostics Corporation, USA). The mutant complex will hybridize less efficiently and melt at a lower temperature, enabling it to be detected even in heterozygotes.
Although the level of α-galactosidase activity in many female heterozygotes is lower than normal and the level of urinary Gb3 is elevated, the only absolutely reliable method of carrier detection is identification of the family mutation in the GLA gene.
Types of mutations/polymorphisms
To date, 363 different mutations have been reported in the GLA gene in the Human Gene Mutation Database (Cardiff, UK: http://www.hgmd.cf.ac.uk/ac/search/119272.html) that are believed to cause Fabry disease. Many have been shown by expression in vitro to abolish or markedly decrease α-galactosidase activity. Others are deduced to be disease causing because they are the only sequence change found in the patient's GLA gene and have not been found in normal controls. The Human Gene Mutation Database contains 245 missense and nonsense mutations, 18 RNA-processing defects, 22 small insertions 58 deletions, 4 indels (a deletion followed by an insertion), 10 gross deletions, 1 gross duplication and 5 complex mutations in which more than one mutational event appears to have occurred. Most are private mutations but some are recurrent, a few of which have been shown by haplotyping to occur in distantly related members of the same family. The majority of the recurrent mutations occur in CpG dinucleotides.
Conclusions
All males who are hemizygous for Fabry disease can be diagnosed by demonstrating a deficiency of α-galactosidase A activity in plasma and/or leukocytes. The mutation in the GLA gene should be determined. Female heterozygotes cannot be diagnosed reliably by enzymatic assay because of skewed inactivation of the X chromosome. Mutation analysis is necessary for the detection of female heterozygotes, and the affected families should be offered genetic counselling. Some individuals have α-galactosidase A activity below the normal reference range due to the presence of a pseudodeficiency allele. This can be confirmed by mutational analysis.
All classic hemizygotes have elevated levels of urinary Gb3, whereas not all hemizygotes with variant forms of Fabry disease have elevated levels. Approximately 95% of female heterozygotes with classic disease have elevated levels of urinary Gb3, whereas the proportion of heterozygotes with variant forms who have elevated Gb3 levels is much lower. The pseudodeficiency allele does not cause elevation of urinary Gb3. The levels of Gb3 in plasma are not so informative, as only 92% and 42% of classic hemizygotes and heterozygotes, respectively, have elevated levels. Prenatal diagnosis of Fabry disease is possible by a combination of enzymatic assay and/or mutation analysis.
Acknowledgements
We acknowledge the collaboration of our colleagues in the Enzyme Diagnostic Laboratory at Great Ormond Street Hospital, London, and in the research laboratories of the Institute of Child Health, London, with whom we have worked for many years and without whom this chapter could not have been written.
References
- 1.
- Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L. Enzymatic defect in Fabry's disease. Ceramidetrihexosidase deficiency. N Engl J Med. 1967;276:1163–7. [PubMed: 6023233]
- 2.
- Kint JA. Fabry's disease: α-galactosidase deficiency. Science. 1970;167:1268–9.
- 3.
- Desnick RJ, Allen KY, Desnick SJ, Raman MK, Bernlohr RW, Krivit W. Fabry's disease: enzymatic diagnosis of hemizygotes and heterozygotes. α-Galactosidase activities in plasma, serum, urine, and leukocytes. J Lab Clin Med. 1973;81:157–71. [PubMed: 4683418]
- 4.
- Beutler E, Kuhl W. Purification and properties of human α-galactosidases. J Biol Chem. 1972;247:7195–200. [PubMed: 4629399]
- 5.
- Desnick RJ, Ioannou YA, Eng CM. α-Galactosidase A deficiency: Fabry disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. 8th edn. New York: McGraw-Hill; 2001. p. 3733–74.
- 6.
- Dean KJ, Sung SS, Sweeley CC. The identification of α-galactosidase B from human liver as an α-N-acetylgalactosaminidase. Biochem Biophys Res Commun. 1977;77:1411–17. [PubMed: 901541]
- 7.
- Schram AW, Hamers MN, Tager JM. The identity of α-galactosidase B from human liver. Biochim Biophys Acta. 1977;482:138–44. [PubMed: 405044]
- 8.
- Desnick RJ, Schindler D. α-N-Acetylgalactosaminidase deficiency: Schindler disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. 8th edn. New York: McGraw-Hill; 2001. p. 3483–505.
- 9.
- Asfaw B, Ledvinova J, Dobrovolny R, Bakker HD, Desnick RJ, van Diggelen OP. et al. Defects in degradation of blood group A and B glycosphingo-lipids in Schindler and Fabry diseases. J Lipid Res. 2002;43:1096–104. [PubMed: 12091494]
- 10.
- Mayes JS, Scheerer JB, Sifers RN, Donaldson ML. Differential assay for lysosomal α-galactosidases in human tissues and its application to Fabry's disease. Clin Chim Acta. 1981;112:247–51. [PubMed: 6263521]
- 11.
- Wang AM, Desnick RJ. Structural organization and complete sequence of the human α-N-acetylgalactosaminidase gene: homology with the α-galactosidase A gene provides evidence for evolution from a common ancestral gene. Genomics. 1991;10:133–42. [PubMed: 1646157]
- 12.
- Lemansky P, Bishop DF, Desnick RJ, Hasilik A, von Figura K. Synthesis and processing of α-galactosidase A in human fibroblasts. Evidence for different mutations in Fabry disease. J Biol Chem. 1987;262:2062–5. [PubMed: 3029062]
- 13.
- Kase R, Bierfreund U, Klein A, Kolter T, Itoh K, Suzuki M. et al. Only sphingolipid activator protein B (SAP-B or saposin B) stimulates the degradation of globotriaosylceramide by recombinant human lysosomal α-galactosidase in a detergent-free liposomal system. FEBS Lett. 1996;393:74–6. [PubMed: 8804427]
- 14.
- Li SC, Kihara H, Serizawa S, Li YT, Fluharty AL, Mayes JS. et al. Activator protein required for the enzymatic hydrolysis of cerebroside sulfate. Deficiency in urine of patients affected with cerebroside sulfatase activator deficiency and identity with activators for the enzymatic hydrolysis of GM1 ganglioside and globotriaosylceramide. J Biol Chem. 1985;260:1867–71. [PubMed: 2981875]
- 15.
- Mills K, Morris P, Lee P, Vellodi A, Waldek S, Young E. et al. Measurement of urinary CDH and CTH by tandem mass spectrometry in patients hemizygous and heterozygous for Fabry disease. J Inherit Metab Dis. 2005;28:35–48. [PubMed: 15702404]
- 16.
- Wherrett JR, Hakomori SI. Characterization of a blood group B glycolipid, accumulating in the pancreas of a patient with Fabry's disease. J Biol Chem. 1973;248:3046–51. [PubMed: 4735571]
- 17.
- Dean KJ, Sweeley CC. Studies on human liver α-galactosidases. I. Purification of α-galactosidase A and its enzymatic properties with glycolipid and oligosaccharide substrates. J Biol Chem. 1979;254:9994–10000. [PubMed: 39940]
- 18.
- Eng CM, Guffon N, Wilcox WR, Germain DP, Lee P, Waldek S. et al. Safety and efficacy of recombinant human α-galactosidase A replacement therapy in Fabry's disease. N Engl J Med. 2001;345:9–16. [PubMed: 11439963]
- 19.
- Schiffmann R, Kopp JB, Austin HA, 3rd, Sabnis S, Moore DF, Weibel T. et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA. 2001;285:2743–9. [PubMed: 11386930]
- 20.
- Blom D, Speijer D, Linthorst GE, Donker-Koopman WG, Strijland A, Aerts JM. Recombinant enzyme therapy for Fabry disease: absence of editing of human α-galactosidase A mRNA. Am J Hum Genet. 2003;72:23–31. [PMC free article: PMC420010] [PubMed: 12471562]
- 21.
- Lee K, Jin X, Zhang K, Copertino L, Andrews L, Baker-Malcolm J. et al. A biochemical and pharmacological comparison of enzyme replacement therapies for the glycolipid storage disorder Fabry disease. Glycobiology. 2003;13:305–13. [PubMed: 12626384]
- 22.
- Ioannou YA, Zeidner KM, Grace ME, Desnick RJ. Human α-galactosidase A: glycosylation site 3 is essential for enzyme solubility. Biochem J. 1998;332(Pt 3):789–97. [PMC free article: PMC1219542] [PubMed: 9620884]
- 23.
- Linthorst GE, Hollak CE, Donker-Koopman WE, Strijland A, Aerts JM. Enzyme therapy for Fabry disease: neutralizing antibodies toward agalsidase alpha and beta. Kidney Int. 2004;66:1589–95. [PubMed: 15458455]
- 24.
- Garman SC, Garboczi DN. The molecular defect leading to Fabry disease: structure of human α-galactosidase. J Mol Biol. 2004;337:319–35. [PubMed: 15003450]
- 25.
- Garman SC, Hannick L, Zhu A, Garboczi DN. The 1.9 Å structure of α-N-acetylgalactosaminidase: molecular basis of glycosidase deficiency diseases. Structure (Camb). 2002;10:425–34. [PubMed: 12005440]
- 26.
- Matsuzawa F, Aikawa SI, Doi H, Okumiya T, Sakuraba H. Fabry disease: correlation between structural changes in α-galactosidase, and clinical and biochemical phenotypes. Hum Genet. 2005;117:317–28. [PubMed: 15924232]
- 27.
- Morgan SH, Rudge P, Smith SJ, Bronstein AM, Kendall BE, Holly E. et al. The neurological complications of Anderson–Fabry disease (α-galactosidase A deficiency) – investigation of symptomatic and presymptomatic patients. Q J Med. 1990;75:491–507. [PubMed: 2167495]
- 28.
- Donnai P, Donnai D, Harris R, Stephens R, Young E, Campbell S. Antenatal diagnosis of Niemann-Pick disease in a twin pregnancy. J Med Genet. 1981;18:359–61. [PMC free article: PMC1048757] [PubMed: 7328615]
- 29.
- Froissart R, Guffon N, Vanier MT, Desnick RJ, Maire I. Fabry disease: D313Y is an α-galactosidase A sequence variant that causes pseudodeficient activity in plasma. Mol Genet Metab. 2003;80:307–14. [PubMed: 14680977]
- 30.
- Yasuda M, Shabbeer J, Benson SD, Maire I, Burnett RM, Desnick RJ. Fabry disease: characterization of α-galactosidase A double mutations and the D313Y plasma enzyme pseudodeficiency allele. Hum Mutat. 2003;22:486–92. [PubMed: 14635108]
- 31.
- Brady RO, Uhlendorf BW, Jacobson CB. Fabry's disease: antenatal detection. Science. 1971;172:174–5. [PubMed: 5547732]
- 32.
- Kleijer WJ, Hussaarts-Odijk LM, Sachs ES, Jahoda MG, Niermeijer MF. Prenatal diagnosis of Fabry's disease by direct analysis of chorionic villi. Prenat Diagn. 1987;7:283–7. [PubMed: 3035532]
- 33.
- Chamoles NA, Blanco M, Gaggioli D. Fabry disease: enzymatic diagnosis in dried blood spots on filter paper. Clin Chim Acta. 2001;308:195–6. [PubMed: 11432396]
- 34.
- Berna L, Asfaw B, Conzelmann E, Cerny B, Ledvinova J. Determination of urinary sulfatides and other lipids by combination of reversed-phase and thin-layer chromatographies. Anal Biochem. 1999;269:304–11. [PubMed: 10222002]
- 35.
- Vance DE, Sweeley CC. Quantitative determination of the neutral glycosyl ceramides in human blood. J Lipid Res. 1967;8:621–30. [PubMed: 6057492]
- 36.
- McCluer RH, Ullman MD, Jungalwala FB. High-performance liquid chromatography of membrane lipids: glycosphingolipids and phospholipids. Methods Enzymol. 1989;172:538–75. [PubMed: 2747542]
- 37.
- Zeidner KM, Desnick RJ, Ioannou YA. Immuno-detection of glycolipid-bound recombinant verotoxin B subunit. Anal Biochem. 1999;267:104–13. [PubMed: 9918661]
- 38.
- Sweeley CC, Dawson G. Determination of glycosphingolipid structures by mass spectrometry. Biochem Biophys Res Commun. 1969;37:6–14. [PubMed: 5346362]
- 39.
- Hemling ME, Yu RK, Sedgwick RD, Rinehart KL Jr. Fast atom bombardment mass spectrometry of glycosphingolipids. Glycosphingolipids containing neutral sugars. Biochemistry. 1984;23:5706–13. [PubMed: 6525335]
- 40.
- Domon B, Costello CE. Structure elucidation of glycosphingolipids and gangliosides using high-performance tandem mass spectrometry. Biochemistry. 1988;27:1534–43. [PubMed: 3365408]
- 41.
- Reinhold BB, Chan S-Y, Chan S, Reinhold VN. Profiling glycosphingolipid structural detail periodate-oxidation, electrospray, collision-induced dissociation and tandem mass-spectrometry. Organic Mass Spectrom. 1994;29:736–46.
- 42.
- Mills K, Johnson A, Winchester B. Synthesis of novel internal standards for the quantitative determination of plasma ceramide trihexoside in Fabry disease by tandem mass spectrometry. FEBS Lett. 2002;515:171–6. [PubMed: 11943216]
- 43.
- Mills K, Eaton S, Ledger V, Young E, Winchester B. The synthesis of internal standards for the quantitative determination of sphingolipids by tandem mass spectrometry. Rapid Commun Mass Spectrom. 2005;19:1739–48. [PubMed: 15909321]
- 44.
- Fauler G, Rechberger GN, Devrnja D, Erwa W, Plecko B, Kotanko P. et al. Rapid determination of urinary globotriaosylceramide isoform profiles by electrospray ionization mass spectrometry using stearoyl-d35-globotriaosylceramide as internal standard. Rapid Commun Mass Spectrom. 2005;19:1499–506. [PubMed: 15880667]
- 45.
- Boscaro F, Pieraccini G, la Marca G, Bartolucci G, Luceri C, Luceri F. et al. Rapid quantitation of globotriaosylceramide in human plasma and urine: a potential application for monitoring enzyme replacement therapy in Anderson–Fabry disease. Rapid Commun Mass Spectrom. 2002;16:1507–14. [PubMed: 12203240]
- 46.
- Mills K, Vellodi A, Morris P, Cooper D, Morris M, Young E. et al. Monitoring the clinical and biochemical response to enzyme replacement therapy in three children with Fabry disease. Eur J Pediatr. 2004;163:595–603. [PubMed: 15243806]
- 47.
- Young E, Mills K, Morris P, Vellodi A, Lee P, Waldek S. et al. Is globotriaosylceramide a useful biomarker in Fabry disease? Acta Paediatr Suppl. 2005;447:51–4. [PubMed: 15895713]
- 48.
- Kitagawa T, Ishige N, Suzuki K, Owada M, Ohashi T, Kobayashi M. et al. Non-invasive screening method for Fabry disease by measuring globotriaosyl-ceramide in whole urine samples using tandem mass spectrometry. Mol Genet Metab. 2005;85:196–202. [PubMed: 15979031]
- 49.
- Whitfield PD, Calvin J, Hogg S, O'Driscoll E, Halsall D, Burling K. et al. Monitoring enzyme replacement therapy in Fabry disease – role of urine globotriaosylceramide. J Inherit Metab Dis. 2005;28:21–33. [PubMed: 15702403]
- 50.
- Roddy TP, Nelson BC, Sung CC, Araghi S, Wilkens D, Zhang XK. et al. Liquid chromatography-tandem mass spectrometry quantification of globotriaosyl-ceramide in plasma for long-term monitoring of Fabry patients treated with enzyme replacement therapy. Clin Chem. 2005;51:237–40. [PubMed: 15514097]
- 51.
- Fuller M, Sharp PC, Rozaklis T, Whitfield PD, Blacklock D, Hopwood JJ. et al. Urinary lipid profiling for the identification of Fabry hemizygotes and heterozygotes. Clin Chem. 2005;51:688–94. [PubMed: 15695328]
- 52.
- Touboul D, Roy S, Germain DP, Baillet A, Brion F, Prognon P. et al. Fast fingerprinting by MALDI-TOF mass spectrometry of urinary sediment glycosphingolipids in Fabry disease. Anal Bioanal Chem. 2005;382:1209–16. [PubMed: 15959771]
- 53.
- Bishop DF, Calhoun DH, Bernstein HS, Hantzopoulos P, Quinn M, Desnick RJ. Human α-galactosidase A: nucleotide sequence of a cDNA clone encoding the mature enzyme. Proc Natl Acad Sci USA. 1986;83:4859–63. [PMC free article: PMC323842] [PubMed: 3014515]
- 54.
- Kornreich R, Desnick RJ, Bishop DF. Nucleotide sequence of the human α-galactosidase A gene. Nucleic Acids Res. 1989;17:3301–2. [PMC free article: PMC317741] [PubMed: 2542896]
- 55.
- Davies JP, Winchester BG, Malcolm S. Sequence variations in the first exon of α-galactosidase A. J Med Genet. 1993;30:658–63. [PMC free article: PMC1016494] [PubMed: 8411052]
- 56.
- Fitzmaurice TF, Desnick RJ, Bishop DF. Human α-galactosidase A: high plasma activity expressed by the −30G>A allele. J Inherit Metab Dis. 1997;20:643–57. [PubMed: 9323559]
- 57.
- Bernstein HS, Bishop DF, Astrin KH, Kornreich R, Eng CM, Sakuraba H. et al. Fabry disease: six gene rearrangements and an exonic point mutation in the α-galactosidase gene. J Clin Invest. 1989;83:1390–9. [PMC free article: PMC303833] [PubMed: 2539398]
- 58.
- Kornreich R, Desnick RJ. Fabry disease: detection of gene rearrangements in the human α-galactosidase A gene by multiplex PCR amplification. Hum Mutat. 1993;2:108–11. [PubMed: 8318986]
- 59.
- Germain D, Biasotto M, Tosi M, Meo T, Kahn A, Poenaru L. Fluorescence-assisted mismatch analysis (FAMA) for exhaustive screening of the α-galactosidase A gene and detection of carriers in Fabry disease. Hum Genet. 1996;98:719–26. [PubMed: 8931708]
- 60.
- Shabbeer J, Robinson M, Desnick RJ. Detection of α-galactosidase a mutations causing Fabry disease by denaturing high performance liquid chromatography. Hum Mutat. 2005;25:299–305. [PubMed: 15712228]
- 61.
- Eng CM, Resnick-Silverman LA, Niehaus DJ, Astrin KH, Desnick RJ. Nature and frequency of mutations in the α-galactosidase A gene that cause Fabry disease. Am J Hum Genet. 1993;53:1186–97. [PMC free article: PMC1682507] [PubMed: 7504405]
- 62.
- Blaydon D, Hill J, Winchester B. Fabry disease: 20 novel GLA mutations in 35 families. Hum Mutat. 2001;18:459. [PubMed: 11668641]
- 63.
- Aoshima T, Sekido Y, Miyazaki T, Kajita M, Mimura S, Watanabe K. et al. Rapid detection of deletion mutations in inherited metabolic diseases by melting curve analysis with LightCycler. Clin Chem. 2000;46:119–22. [PubMed: 10620581]
- Plasma globotriaosylsphingosine as a biomarker of Fabry disease.[Mol Genet Metab. 2010]Plasma globotriaosylsphingosine as a biomarker of Fabry disease.Togawa T, Kodama T, Suzuki T, Sugawara K, Tsukimura T, Ohashi T, Ishige N, Suzuki K, Kitagawa T, Sakuraba H. Mol Genet Metab. 2010 Jul; 100(3):257-61. Epub 2010 Apr 1.
- Phenotypical characterization of α-galactosidase A gene mutations identified in a large Fabry disease screening program in stroke in the young.[Clin Neurol Neurosurg. 2013]Phenotypical characterization of α-galactosidase A gene mutations identified in a large Fabry disease screening program in stroke in the young.De Brabander I, Yperzeele L, Ceuterick-De Groote C, Brouns R, Baker R, Belachew S, Delbecq J, De Keulenaer G, Dethy S, Eyskens F, et al. Clin Neurol Neurosurg. 2013 Jul; 115(7):1088-93. Epub 2012 Dec 4.
- Urinary globotriaosylceramide excretion correlates with the genotype in children and adults with Fabry disease.[Mol Genet Metab. 2008]Urinary globotriaosylceramide excretion correlates with the genotype in children and adults with Fabry disease.Auray-Blais C, Cyr D, Ntwari A, West ML, Cox-Brinkman J, Bichet DG, Germain DP, Laframboise R, Melançon SB, Stockley T, et al. Mol Genet Metab. 2008 Mar; 93(3):331-40. Epub 2007 Nov 26.
- Review Anderson-Fabry disease: developments in diagnosis and treatment.[Acta Clin Croat. 2012]Review Anderson-Fabry disease: developments in diagnosis and treatment.Kes VB, Cesarik M, Zavoreo I, Madzar Z, Demarin V. Acta Clin Croat. 2012 Sep; 51(3):411-7.
- Review Early therapeutic intervention in females with Fabry disease?[Acta Paediatr. 2008]Review Early therapeutic intervention in females with Fabry disease?Hughes DA. Acta Paediatr. 2008 Apr; 97(457):41-7.
- Biochemical and genetic diagnosis of Fabry disease - Fabry DiseaseBiochemical and genetic diagnosis of Fabry disease - Fabry Disease
Your browsing activity is empty.
Activity recording is turned off.
See more...